| Informations
sur la CMT |
|||
|
La maladie de Charcot-Marie-Tooth, ou CMT, est l’une des maladies neurologiques les plus fréquentes. La maladie de Charcot-Marie-Tooth (CMT en abrégé) doit son nom aux trois médecins qui l’ont décrite en 1886 : deux français CHARCOT et MARIE et un anglais TOOTH.
Elle est parfois appelée Amyotrophie
neurogène. Cette maladie est une neuropathie héréditaire
sensitivomotrice qui n’affecte pas l’espérance de vie et n’entraîne
pas de retard mental. Elle touche indifféremment l’homme ou la femme. |
|
||
|
La CMT est une maladie génétique, héréditaire, elle est due à une anomalie chromosomique transmise par l’un des parents qui est lui-même malade, parfois sans signes cliniques (mode dominant) ou par les deux parents, porteurs sains (mode récessif). Parfois il s’agit d’une mutation génétique lorsqu’il n’existe aucun cas connu dans la famille.
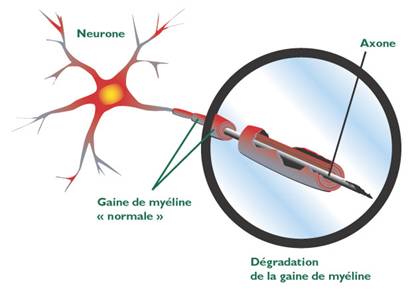
Schématiquement, la CMT est liée à l’atteinte de la gaine des
neurones des nerfs périphériques, ce qui perturbe la conduction de l’influx
nerveux. Elle entraîne des troubles de la marche et une déformation
fréquente des pieds. Cette maladie peut se déclarer assez tard et
même à l’âge adulte. En général, la CMT évolue lentement mais
elle peut aussi progresser par poussées en particulier à l’adolescence,
quelquefois lors d’une grossesse. La prise de vitamine B6 (Pyridoxine)
peut aggraver la maladie. La gravité de l’atteinte est différente d’une
personne à l’autre et rien ne permet de prédire l’évolution. Le
degré de handicap peut aller d’une simple gêne à la marche, jusqu’à
l’usage d’un fauteuil roulant (peu fréquent : environ 10% des cas). |
|||
|
Les principaux types de CMT sont les suivants :
Type 1
: Atteinte de la myéline, Transmission autosomique dominante
Type 2 : Atteinte de l'axone
Type 4
: Atteinte de la myéline, Transmission autosomique récessive
Type X
: Transmission dominante liée à l'X
|
|
||